Biological Sciences
Published
Author Stephen Turner
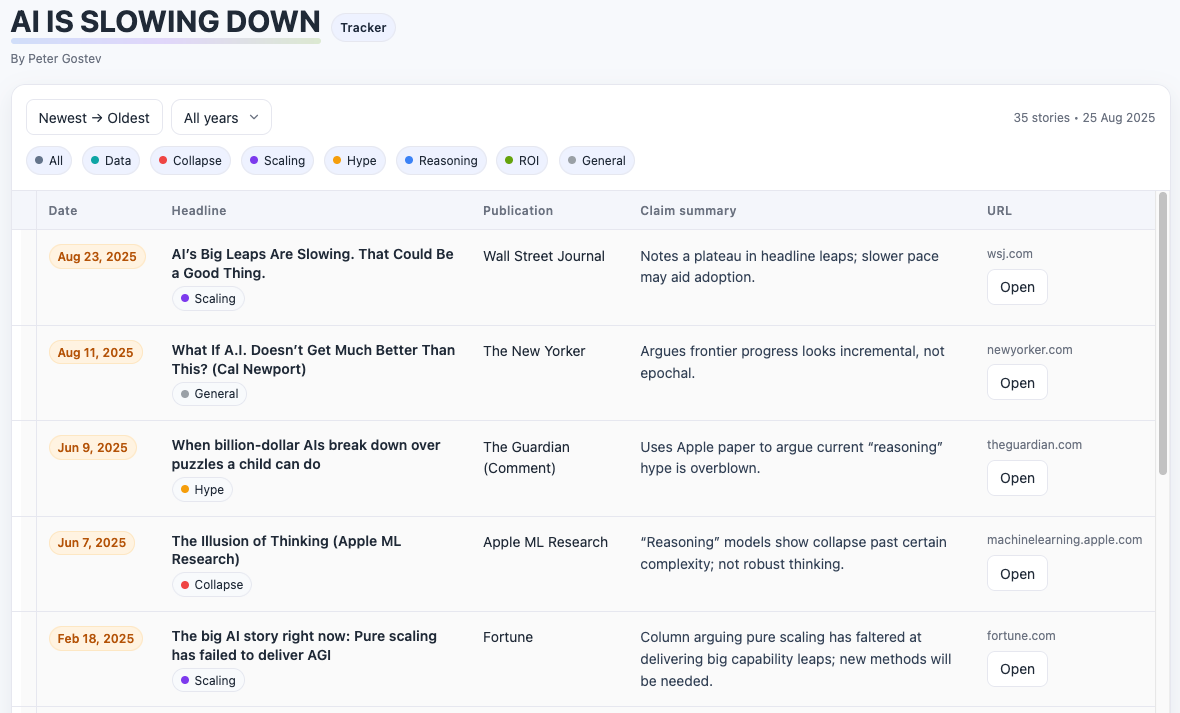
Happy Friday, colleagues. August has flown by at warp speed. I started a new job, and my backlog of (semi-) pleasure reading grows longer. This is my regular attempt to close out my browser tabs I’ve accumulated over the past week with blog posts, podcasts, papers, etc. in AI, data science, genomics, public health, programming, scicomm, and other miscellany.