BiologiaInglese
Pubblicato
Autore Stephen Turner

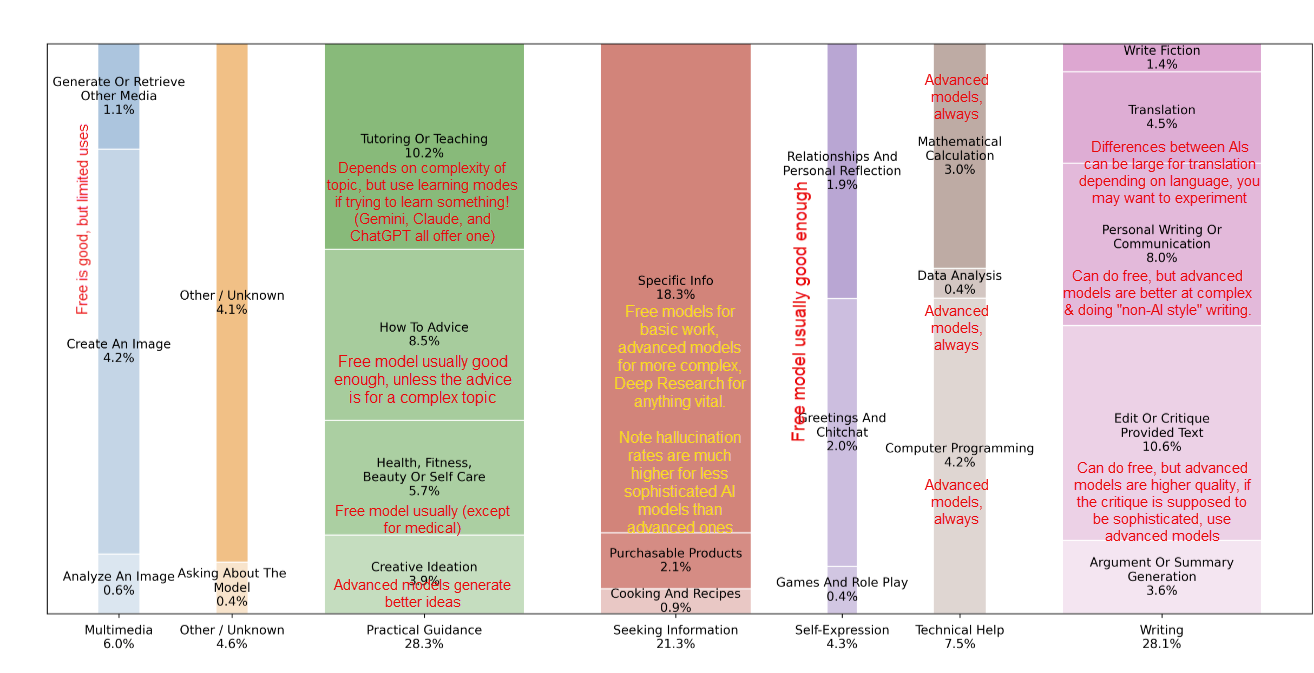
The Bioconductor 3.22 release is now available. It includes 2,361 software packages, 435 experiment data packages, 926 annotation packages, 29 workflows, and 6 books. This cycle adds 59 new software packages, 6 new experiment data packages, 2 new annotation packages, and 1 new book, along with many updates across the existing ecosystem. A few themes stand out for people working in genomics, single-cell transcriptomics, and spatial omics.