Crystal_structure_miningInteresting ChemistryAngleMetalSubtended AngleCiencias QuímicasInglés
Publicado
Autor Henry Rzepa
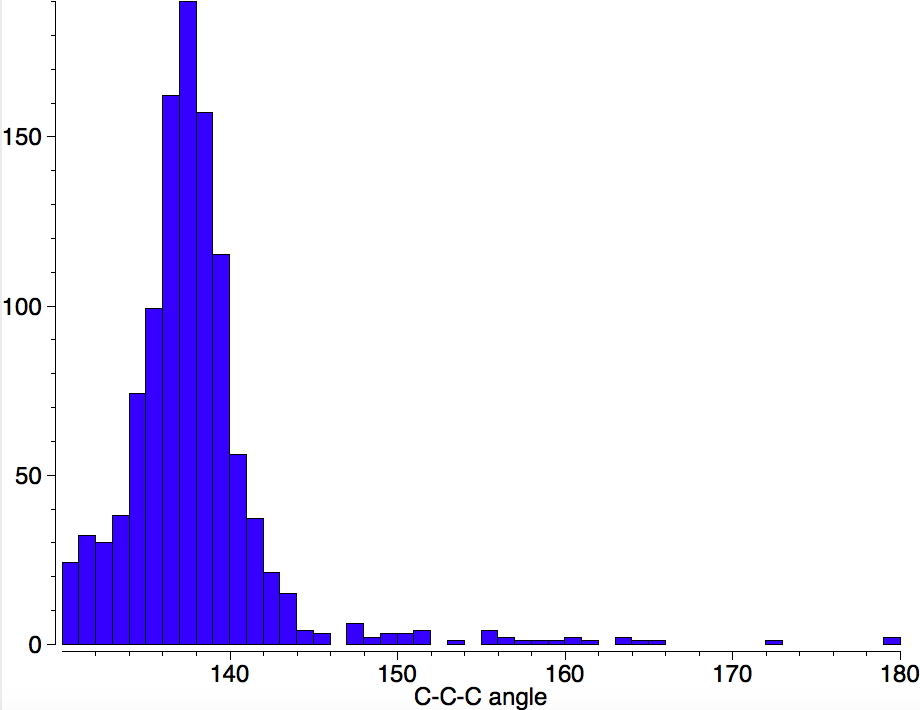
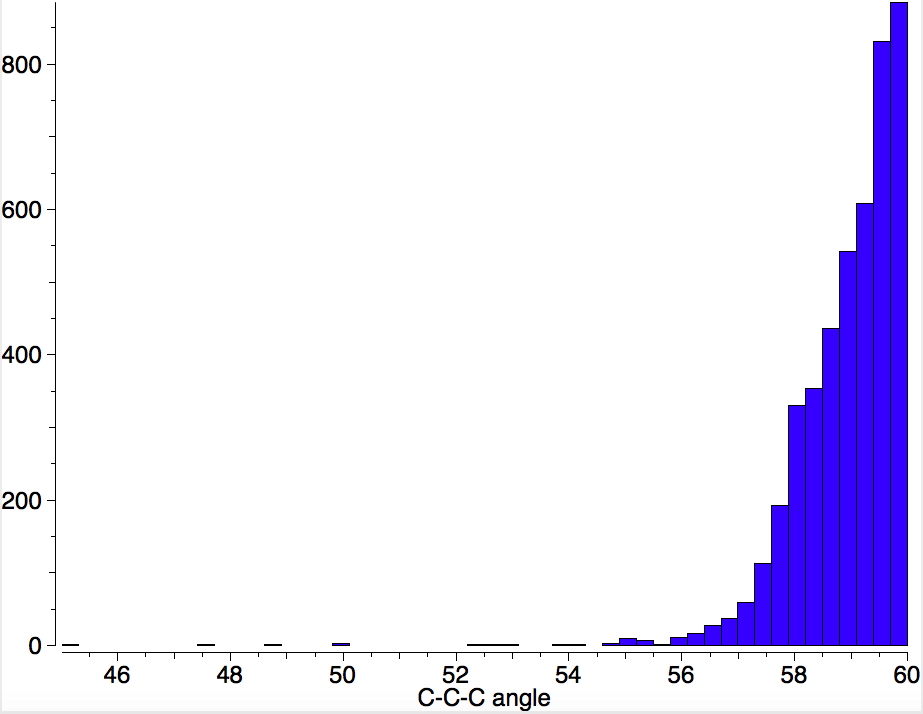
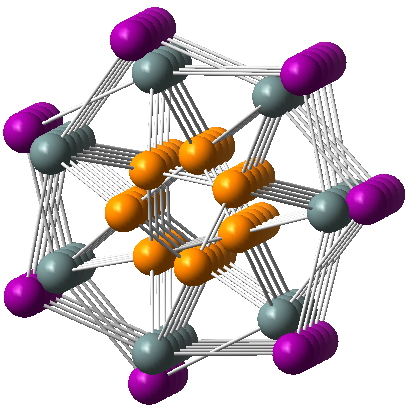
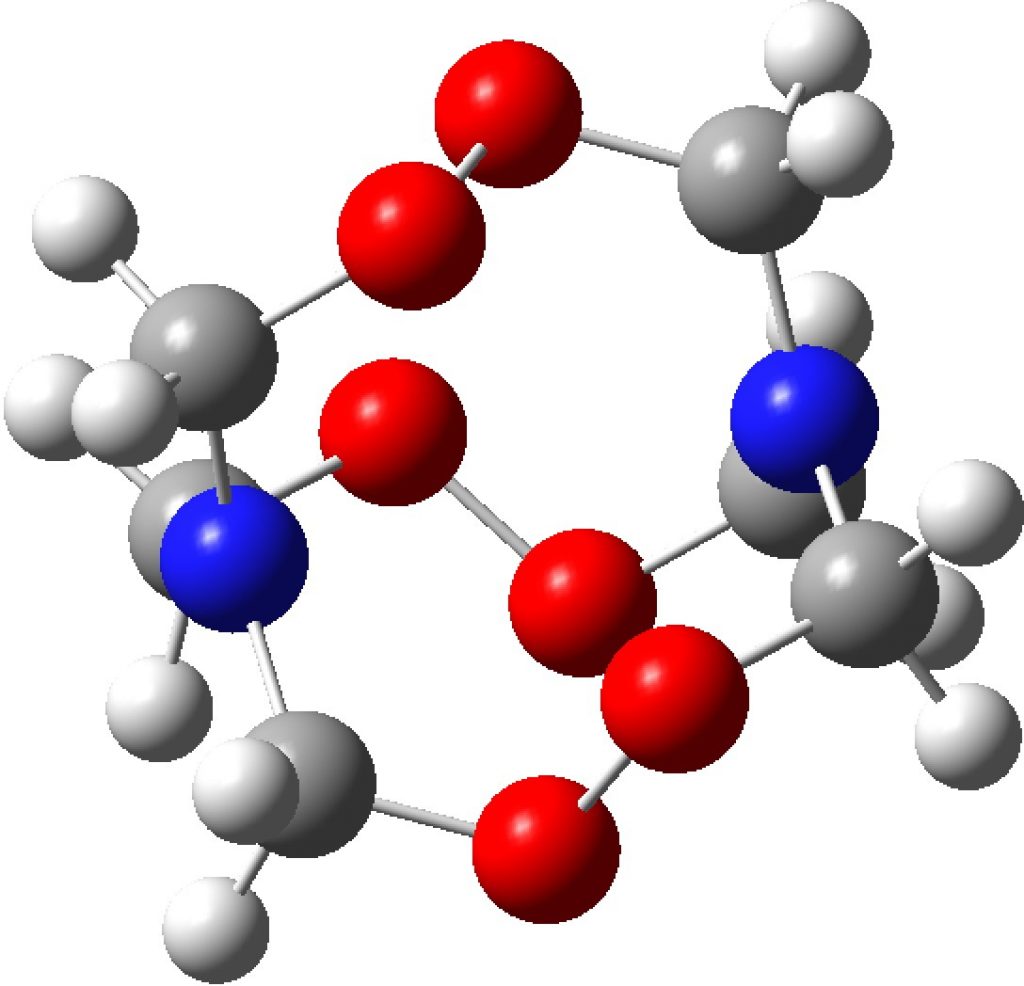
I am now inverting the previous question by asking what is the largest angle subtended at a chain of three connected 4-coordinate carbon atoms? Let’s see if further interesting chemistry can be unearthed.